
WebAUGUSTUS Training Tutorial
This website explains step-by-step how to use the AUGUSTUS training web server application to train AUGUSTUS parameters for you individual species of interest. You find a similar tutorial on how to predict genes with pre-trained AUGUSTUS parameters here (click).
Functionalities of the AUGUSTUS training web server application are (with a single run):
- The generation of training gene structures (if no training gene structures are available)
- Optimization of AUGUSTUS parameters according to the training gene structures
- Prediction of genes in a supplied genome file with the newly optimized AUGUSTUS parameters. Genes will be predicted ab initio and with hints (the latter only if a cDNA file is provided).
Before submitting a training job for your species of interest, please check whether parameters have already been trained and have been made publicly available for your species at our species overview table
1 - Job Submission in general
1.1 - Finding the training submission form
1.2 - Filling in general job data
1.2.1 - E-mail address
1.2.1.1 - What your e-mail address is used for
1.2.2 - Species name
1.2.2.1 - The species name format
1.2.2.2 - Purpose of entering a species name
1.2.3 - Genome file
1.2.3.1 - Genome file format
1.2.3.2 - Genome file upload options
1.2.3.3 - What the genome file is used for
1.3 - Optional obligatory fields
1.3.1 - cDNA file
1.3.1.1 - cDNA file format
1.3.1.2 - cDNA file upload options
1.3.1.3 - What cDNA files are used for
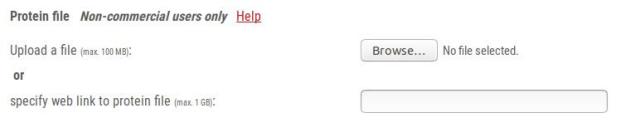
1.3.2 - Protein file
1.3.2.1 - Protein file format
1.3.2.2 - Protein file upload options
1.3.2.3 - What the protein file is used for
1.3.3 - Training gene structure file
1.3.3.1 - Training gene structure file format
1.3.3.1.1 - Training gene structure file in genbank format
1.3.3.1.2 - Training gene structure file in gff format
1.3.3.2 - What training gene structure files are used for
1.4 - Verification that you are a human
1.5 - The submit button
1.6 - Example data files
2 - What happens after submission
2.1 - Submission duplication
2.2 - Errors during training
3 - Training Results
3.1 - The AutoAug.log file
3.2 - The AutoAug.err file
3.3 - The parameters.tar.gz archive
3.4 - The training.gb.gz file
3.5 - Gene prediction archives
References
The pipeline invoked by submitting a job to the AUGUSTUS training web server application is complex. The following image gives an overview of processes that will possibly be invoked - depending on the input file combination:
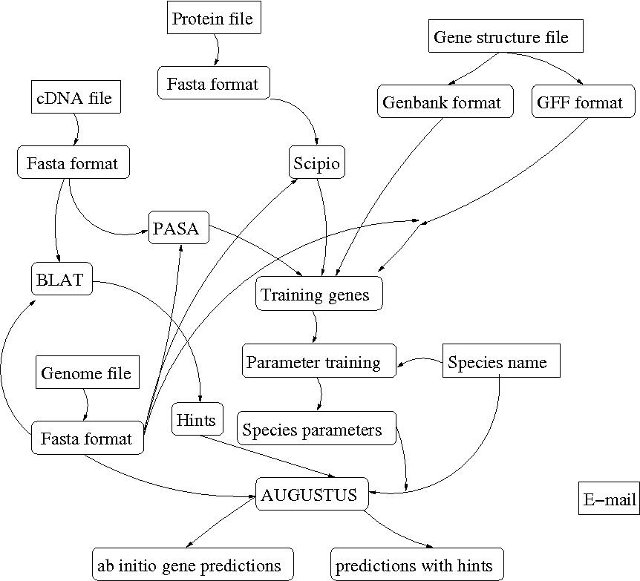 |
The sharp-edge fields are input values of the submission form, the round edged fields are background processes/applied software tools/file formats/result files.
The input fields of the AUGUSTUS training web server application form are: E-mail, Species name, Genome file, cDNA file, Protein file and Training gene structure file. The actual processes invoked by job submission depend on the combination of input files.
- {genome file, cDNA file}
In this case, the cDNA file is used to create training gene structures with PASA [1]. If cDNA end quality is sufficient, also a UTR training set will be created (this is currently the only possibility to train UTR parameters using this web server application). After parameters have been trained using the so created gene structure file, the cDNAs will additionally be used to create hints. Hints are extrinsic evidence for gene structures that are used during gene prediction with hints. The mapping tool BLAT [2] is used during hint generation. BLAT is available for academic, personal and non-profit use on our web server, only! Finally, AUGUSTUS [4] is used to predict genes in the genome file ab initio and with hints. - {genome file, protein file}
In this case, the protein file is used to create a training gene set using Scipio. Scipio [3] uses BLAT [2]. BLAT is available for academic, personal and non-profit use on our web server, only! After parameter optimization, AUGUSTUS [4] is used to predict genes in the genome sequence ab initio. - {genome file, gene structure file}
In this case, the gene structure file is used as a training gene set. Gene structure files can be provided in two different formats: genbank format and gff format. If a genbank file is submitted, there is no dependency between training gene structure file and genome file. Parameters are then optimized based on the genbank training gene structure file. If a gff file is submitted, the gff must comply with the genome file entries. Training gene sequences are extraced from the genome file prior parameter optimization. Finally, AUGUSTUS [4] is used for ab initio gene prediction. The submission of training gene structure file in combination with a genome file is open to all users! - {genome file, cDNA file, protein file}
In this case, the protein file will be used to create a training gene set using Scipio [3]. No UTR training set will be created. cDNA sequences will be used as evidence for gene prediction, only. Since both Scipio and hint generation employ BLAT [2], this file combination is available for academic, personal and non-profit use on our web server, only! Finally, AUGUSTUS [4] is used to predict genes in the genome file ab initio and with hints. - {genome file, cDNA file, gene structure file}
In this case, the gene structure file is used as a training gene set. cDNA sequences will be used as evidence for prediction in form of hints that are generated with the help of BLAT. Since hint generation employs BLAT [2], this file combination is available for academic, personal and non-profit use on our web server, only! Finally, AUGUSTUS [4] is used to predict genes in the genome file ab initio and with hints.
In the following, you find detailed instructions for submitting an AUGUSTUS training job.
You find the WebAUGUSTUS training submission form by clicking on the following link in the left side navigation bar:
 |
This section describes fields that should be filled in for every job submission, i.e. fields that are obligatory (except for the email address, which is optional but strongly recommended).
At first, we recommend that you enter a valid e-mail address:
 |
It is possible to run WebAUGUSTUS without giving an e-mail address but here are some reasons why we recommend supplying an e-mail address:
- Unlike many other bioinformatics web services, the AUGUSTUS web server application is not an implementation of a fail-safe procedure. Particularly the assembly of a training gene set from extrinsic data (ESTs and protein sequences) and a genome sequence may not always work perfectly. Our pipeline may issue warnings or errors, and sometimes, we need to get some feedback from you via e-mail in order to figure out what is the problem with your particular input data set.
- In addition, training AUGUSTUS is rather time consuming process that may take up to several weeks (depending on the input data). It may be more convenient to receive an e-mail notification about your job having finished, than checking the status page over and over, again
We use your e-mail address for the following purposes:
- Confirming your job submission
- Confirming successful file upload (for large files via ftp/http link)
- Notifying you that your job has finished
- Informing you about any problems that might occur during your particular AUGUSTUS training job
We do not use your e-mail address to send you any spam, i.e. about web service updates. We do not share your e-mail address with any third parties. Please read our Data Privacy Protection declaration.
 |
The species name must be a string of at most 30 characters length. It is not allowed to contain spaces.
The species name is the name of the species for whose genome you want to train AUGUSTUS. The species name is an obligatory parameter. Considering that AUGUSTUS training is such a time consuming process, our objective is to know the names of species for which AUGUSTUS was trained in order to make the trained parameters available to the public so that others who are interested in the same species as you do not have to rerun the training process. (You will be asked for permission before your parameter set is shared.)
However, if you do not want to reveal the true species name, you may use any other string shorter than 30 characters as a species name.
The genome file is an obligatory file for training AUGUSTUS.
The genome file must contain the genome sequence in (multiple) fasta format. Every unique header begins with a >. The sequence must be DNA. Allowed sequence characters: A a T t G g C c H h X x R r Y y W w S s M m K k B b V v D d N n. (Internally, AUGUSTUS will interpret everything that is not A a T t C c G g as an N!) Empty lines are not allowed. If they occur, they will automatically be removed by the webserver application. White spaces in the sequence header might cause problems if the first word after the leading character > is identical for several fasta entries. We generally recommend short, unique, non-white-space containing fasta headers.WebAUGUSTUS does have a strict limit for character per line for FASTA format files. Disobeying this restriction might cause memory issues on our server. We recommend to format sequences in FASTA files submitted to WebAUGUSTUS with a unix linebreak after 80 characters.
Correct file format example:
>Chr.1
CCTCCTCCTGTTTTTCCCTCAATACAACCTCATTGGATTATTCAATTCAC
CATCCTGCCCTTGTTCCTTCCATTATACAGCTGTCTTTGCCCTCTCCTTC
TCTCGCTGGACTGTTCACCAACTCTCAGCCCGCGATCCCAATTTCCAGAC
AACCCATCTTATCAGCTTGGCCACGGCCTCGACCCGAACAGACCGGCGTC
CAGCGAGAAGAGCGTCGCCTCGACGCCTCTGCTTGACCGCACCTTGATGC
TCAAGACTTATCGCGATGCCAAGAAGCGTCTCATCATGTTCGACTACGA
>Chr.2
CGAAACGGGCACCTATACAACGATTGAAACCATTATTCAAGCTCAGCAAG
CGTCTATGCTAGCGGTTATTGCGAGCACTTCAGCGGTTGCTACTACGACT
ACTACTTGATAAATGAAACGGCTATAAAAGAGGCTGGGGCAAAAGTATGT
TAGTTGAAGGGTGACCTGAACGATGAATCGGTCGAATTTTTTATTGGCAG
AGGGAAGGTAGGTTTACTCAATTTAGTTACTTCTAGCCGTTGATTGGAGG
AGCGCAAGCGACGAGGAGGCTCATCGGCCGCCCGCGGAAAGCGTAGTCT
TACACGGAAATCAACGGCGGTGTCATAAGCGAG
>Chr.3
.....
|
The maximal number of scaffolds allowed in a genome file is 250000. If your file contains more scaffolds, please remove all short scaffolds. For training AUGUSTUS, short scaffolds are worthless because no complete training genes can be generated from them. In terms of prediction, it is possible to predict genes in short scaffolds. However, those genes will in most cases be incomplete and probably unreliable.
Besides plain fasta format, our server accepts gzipped-fasta format for genome file upload. You find more information about gzip at the gzip homepage. Gzipped files have the file ending *.gz.
The AUGUSTUS training web server application offers two possiblities for transferring the genome file to the server: Upload a file and specify a web link to file.
 |
- For small files, please click on the Choose File or Browse-button and select a file on your harddrive.
If you experience a Connection timeout (because your file was too large for this type of upload - the size is browser dependent), please use the option for large files! - Large files can be retrieved from a public web link. Deposit your sequence file at a http or ftp server and specify the valid URL to your sequence file in the training submission form. Our server will fetch the file from the given address upon job submission. (File size limit: currently 1 GB. Please contact us in case you want to upload a bigger genome file, links to dropbox are not supported by WebAUGUSTUS.) You will be notified by e-mail when the file upload from web-link is finished (i.e. you can delete the file from the public server after you received that e-mail).
You cannot do both at the same time! You must either select a file on your harddrive or give a web link!
The genome file can be used for two purposes:
- Training gene structure generation (if no training gene structure file was submitted or if the training gene structure was supplied in gff format)
- Target sequence for gene prediction with the new AUGUSTUS parameters (always)
This section describes a number of fields from which at least one must be specified for training AUGUSTUS.
This feature is only for personal, academic, and non-profit use as this is required by the BLAT license.
 |
The cDNA file is a multiple fasta DNA file that contains e.g. ESTs or full-length cDNA sequences. Allowed sequence characters: A a T t G g C c H h X x R r Y y W w S s M m K k B b V v D d N n U u. Empty lines are not allowed and will be removed from the submitted file by the webserver application. An example for correct cDNA file format is given at 1.2.3.1 - Genome file format.
It is currently possible to submit assembled RNA-seq transcripts instead of or mixed with ESTs as a cDNA/EST file. However, you should be aware that the success of creating training genes with assembled RNA-seq data depends very much on the assembly quality, and that RNA-seq files are often much bigger than EST or cDNA files, which increases runtime of a training job. In order to keep runtime of your training job as low as possible, you should remove all assembled RNA-seq transcripts from your file that do not map to the submitted genome sequence. (In principle, this holds true for EST and cDNA files, too, but there, the problem is not as pronounced due to a smaller number of sequences.)
It is currently not allowed to upload RNA-seq raw sequences. (We filter for the average length of cDNA fasta entries and may reject the entire training job in case the sequences are on average too short, i.e. shorter than 400 bp.)
Besides plain fasta format, our server accepts gzipped-fasta format for cDNA file upload. You find more information about gzip at the gzip homepage. Gzipped files have the file ending *.gz. The maximal supported file size is 1 GB.
There are two options for cDNA file upload: upload from your local harddrive, or upload from a public http or ftp server. Please see 1.2.3.2 - Genome file upload options for a more detailed description of upload options.
The cDNA file can be used for two purposes by the AUGUSTUS training web server application:
- for generating training gene structures (if no protein file and no training gene structure file are specified)
- for generating extrinsic evidence for gene structures in the gene prediction process (always)
 |
The protein file is a multiple fasta file that contains protein sequences, e.g. from a closely related species. Allowed sequence characters: A a R r N n D d C c E e Q q G g H h I i L l K k M m F f P p S s T t W w Y y V v B b Z z J j X x. Empty lines are not allowed but will simply be removed from the file by the webserver application.
Correct file format example:
>protein1
maaaafgqlnleepppiwgsrsvdcfekleqigegtygqvymakeiktgeivalkkirmd
neregfpitaireikilkklhhenvihlkeivtspgrdrddqgkpdnnkykggiymvfey
mdhdltgladrpglrftvpqikcymkqlltglhychvnqvlhrdikgsnllidnegnlkl
adfglarsyshdhtgnltnrvitlwyrppelllgatkygp
>protein2
neregfpitaireikilkklhhenvihlkeivtspgrdrddqgkpdnnkykggiymvfey
mdhdltgladrpglrftvpqikcymkqlltglhychvnqv
>protein3
...
|
There are two options for protein file upload: upload from your local harddrive, or upload from a public http or ftp server. Please see 1.2.3.2 - Genome file upload options for a more detailed description of upload options.
The protein file is used for generating training gene structures by mapping the protein sequences against the supplied genome sequence. You may e.g. upload a file with protein sequences from a closely related species in order to obtain training genes and AUGUSTUS parameters for the new species whose genome you uploaded.
 |
Training gene structure files can be submitted in two different formats: Genbank format or gff format.
Gene structures in genbank format must contain the coding sequence parts and flanking regions. Flanking regions are important because AUGUSTUS is supposed to differentiate between genes and intergenic regions. The length of flanking regions depends on the length of genes in the target genome. In our pipeline, flanking regions are set to the average gene length (exceptionally applying the extreme limits between 1000 and 10000 nt). It is very important to make sure that the flanking regions do not contain any other protein coding gene parts, i.e. we recommend to trim flanking regions in a way that will exclude other CDS parts.
Correct file format example (condensed view, the three dots represent further lines of sequences):
LOCUS Chr.1_1-159458 159458 bp DNA
FEATURES Location/Qualifiers
source 1..159458
CDS complement(join(2421..2655,3858..4005,4080..4235,5569..5857
,10316..10534,155240..155458))
/gene="1474336"
BASE COUNT 49195 a 29117 c 28985 g 49950 t 2211 n
ORIGIN
1 aaaatacatc acaatacatt taattcactt tccatcatcg agattaacga aaattattta
61 aaatatcgaa gatgaaaata tcctcaagat gatactgaac ggctaagaaa aatacatcac
121 acaactttaa ttcattttcc atcatcgaga ttaacgaaaa gaaaaaattt taactcccta
...
159301 atacgccacc aggtatttcg cctgattgtt cctcgaatat cttctctctc tctatatata
159361 tatatattac ttggcacgat aatcgtcgaa tcgttattta taaattgctt catctatcgc
159421 gatatttttg caacaactct cgcttttctc tttccatt
//
LOCUS Chr.1_313992-323129 9138 bp DNA
FEATURES Location/Qualifiers
source 1..9138
CDS join(4001..4048,4989..5138)
/gene="194551"
BASE COUNT 2829 a 1502 c 1750 g 2948 t 109 n
ORIGIN
1 ttttccttct ttcttttttt tttatttaca ttaatgagaa ttttcgcaaa tatttcatcg
61 ctgccatcct tttttttcct cgacgtcaat cacgcgacac atttgttaga gaaatggatt
121 ttaatcttga aaaaagaaaa atacaaatgc caacgcattt caaatccttt cctattatta
...
9001 tcaacgaaac aaataattgc ttcacaaaat atcgcacgta acaacaatat agacttcaat
9061 attcaacaat tcttttcctt tatacacaaa gatacacaaa atataaaagt tttaatactt
9121 caacttcaac gaaacagg
//
|
If you want to train UTRs, you have to additionally incorporate mRNA information in your genbank file.
Correct file format example (including UTR training):
LOCUS scf7180001240730_g20 526 bp DNA
FEATURES Location/Qualifiers
source 1..526
mRNA 99..125
CDS 99..99
BASE COUNT 164 a 99 c 68 g 195 t
ORIGIN
1 gtgacggagc ccaaggacga gcccgtgccc tcagagccca cgtccgacgt gaggcccgcg
61 ccagcgcccc tcccgccgcc cgtcgcagcc actgcttaga ctttactaat ataaacattg
121 aaaatatttt gtgttttatt tccaatcatt gaattataat cctattataa tataactaac
181 attcgtaatt ttacaaaata actatgcaaa ttattttgta ttttcgtttt aaattatact
241 tttcatataa atttctacaa atcttattca agaccataag tatccgctcg ctctacttcg
301 ggcatttcct ttatttatat cttatttgac ttattttgat tatttaggct tatgttttcg
361 atactattga aaacagaaaa taatttcata taattaataa tatattttca attaatatat
421 ttaacaaata tttgtatagt tcaagcggac aaatccgttc ccatagtatt tatataaatt
481 ttaatttaga gtaataacag tttgctgtat tgttgtagtc aaatac
//
LOCUS scf7180001240751_g30 876 bp DNA
FEATURES Location/Qualifiers
source 1..876
mRNA complement(401..777)
CDS complement(777..777)
BASE COUNT 300 a 136 c 116 g 324 t
ORIGIN
1 aatgtaggaa aatgaaatat ttatttaaat tgttattatc acttcttcgc tctagtgtct
61 tggcaaagcg cggcgttgag ttcagcctct cacacgcaat gcctccagaa ttcggcgaaa
121 tgtgggggac agagtgtatt aacactaagt tccctcagcc acgactggtg aaattatata
181 ttcagtttgt atactattac tcatgcaaac acttcatcat actttcactc aatcagtaaa
241 gcataatatt ttatttaata ttgtttatca atactatttc cttgttgtta aatattattt
301 tatttattat attaaattaa aatgtcaaaa ttaaaagtag gtgatgattt attactatct
361 tttctatcca agaaaaaaaa gacacactga aacaattgta atttttgtta tgtttttatt
421 acttaatatt attataaaaa tttgtaaata cgaaataaaa tagatagacg taataatatt
481 tatttgttag ttaataataa taatgataat tacgaaagat acaagaaata tgcataaatg
541 agtgttatat tatgtatttt atgagaatat aaatataaaa actgtcattg attatatttt
601 ctaaatactt tcattttatg gcttgctggc ttttcaattt ccttatgttt cagcttttca
661 ctcaatagag cgaaaccttc atcgacatgt aagccaatag aacaattaca aactaacttt
721 attacatcag tcttttcatt tctttaagct tcaggcaaat atcatctaaa tgcctttcaa
781 ctcgctacta acatcgcgtc gttatataaa tcagtgtata cggaattaaa cctgtcatgt
841 ctcttgcaag acgtgtctgc tgttgtcacg cacaca
//
|
Training gene structure in gff format must comply with the fasta entry names of the genome file.
In general, gff format must contain the following columns (The columns are separated by tabulators):
- The sequence names must be found in the fasta headers of sequences in the genome file.
- The source tells with which software/process the gene structure was generated (you can fill in whatever you like).
- The feature may for AUGUSTUS training be CDS, 5'-UTR or 3'-UTR.
- Start is the beginning position of the line's feature, counting the first position of a sequence as position 1.
- Stop position, must be at least as large as start position.
- The score must be a number but the number is irrelevant to our web server applications.
- The strand denotes whether the gene is located on the forward (+) or on the reverse (-) strand.
- Frame is the reading frame, can be denoted as '.' if unknown or irrelevant. For exonpart and exon this is as defined as follows: On the forward strand it is the number of bases after (begin position 1) until the next codon boundary comes (0, 1 or 2). On the reverse strand it is the number of bases before (end position + 1) the next codon boundary comes (0, 1 or 2).
- Attribute contains a transcript identifier. All gff-entries belonging to one transcript must contain the same transcript identifier in the last column.
Correct file format example (without UTR):
Chr.1 mySource CDS 1767 1846 1.000 - 0 transcript_id "1597_1" Chr.1 mySource CDS 1666 1709 1.000 - 1 transcript_id "1597_1" Chr.1 mySource CDS 1486 1605 1.000 - 2 transcript_id "1597_1" Chr.1 mySource CDS 1367 1427 1.000 - 2 transcript_id "1597_1" Chr.1 mySource CDS 1266 1319 1.000 - 1 transcript_id "1597_1" Chr.1 mySource CDS 1145 1181 1.000 - 1 transcript_id "1597_1" Chr.1 mySource CDS 847 1047 1.000 - 0 transcript_id "1597_1" Chr.2 mySource CDS 9471 9532 1.000 + 0 transcript_id "1399_2" Chr.2 mySource CDS 9591 9832 1.000 + 1 transcript_id "1399_2" Chr.2 mySource CDS 9885 10307 1.000 + 2 transcript_id "1399_2" Chr.2 mySource CDS 10358 10507 1.000 + 2 transcript_id "1399_2" Chr.2 mySource CDS 10564 10643 1.000 + 2 transcript_id "1399_2" |
Correct file format example (with UTR):
Chr.1 mySource 5'-UTR 277153 277220 45 + . transcript_id "g22472.t1"; gene_id "g22472"; Chr.1 mySource CDS 277221 277238 1 + 0 transcript_id "g22472.t1"; gene_id "g22472"; Chr.1 mySource CDS 278100 278213 1 + 0 transcript_id "g22472.t1"; gene_id "g22472"; Chr.1 mySource CDS 278977 279169 1 + 0 transcript_id "g22472.t1"; gene_id "g22472"; Chr.1 mySource CDS 279630 279648 0.94 + 2 transcript_id "g22472.t1"; gene_id "g22472"; Chr.1 mySource CDS 279734 279768 0.94 + 1 transcript_id "g22472.t1"; gene_id "g22472"; Chr.1 mySource CDS 280307 280344 1 + 2 transcript_id "g22472.t1"; gene_id "g22472"; Chr.1 mySource 3'-UTR 280345 280405 78 + . transcript_id "g22472.t1"; gene_id "g22472"; |
While the cDNA and protein file upload options fulfill the purpose of training gene generation in context with a genome sequence file, the gene structure file upload form provides a flexible interface to users who generated training gene structures externally instead of using our webserver application for this purpose. Supplied training gene structures are directly used for optimizing species-specific AUGUSTUS parameters.
If the training gene structure file is in gff-format, sequence names in the gff file have to match fasta headers in the genome file. If the training gene structure file is in genbank format, no direct dependency between training gene structure file and genome file exists, i.e. you could submit training genes that were generated from a different chromosome than the genome file that you submit.
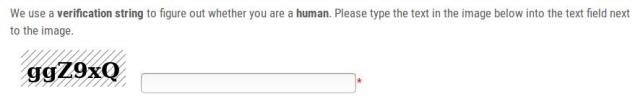 |
Trying to avoid abuse of our web server application through bots, we implemented a captcha. The captcha is an image that contains a string. You have to type the string from the image into the field next to the image.
 |
After filling out the appropriate fields in the submission form, you have to click on the button that says "Start Training" at the bottom of the page. It might take a while until you are redirected to the status page of your job. The reason is that we are checking various file formats prior job acceptance, and that the transfer of files from your local harddrive to our server might take a while. Please be patient and wait until you are redirected to the status page! Do not click the button more than once (it won't do any harm but it also doesn't speed up anything).
In the following, we provide some correctly formatted, compatible example data files:
1.6.1 - Genome and ESTs
http://bioinf.uni-greifswald.de/webaugustus/examples/chr1to3.fa - This file may be used as a Genome file. It contains the first three chromosomes of Mus musculus from GenBank (modified headers).
http://bioinf.uni-greifswald.de/webaugustus/examples/estsChr1to3.fa - This file may be used as a cDNA file. It contains EST sequences of Mus musculus from GenBank (modified headers).
1.6.2 - Genome and Proteins
http://bioinf.uni-greifswald.de/webaugustus/examples/chr1to6.fa - This file may be used as a Genome file. It contains the first six chromosomes of Mus musculus from GenBank (modified headers).
http://bioinf.uni-greifswald.de/webaugustus/examples/rattusProteinsChr1to6.fa - This file may be used as a Protein file. It contains proteins of Rattus norvegicus from GenBank (modified headers) that map to chromosomes 1 to 6.
1.6.3 - Genome and Gene Structure File
http://bioinf.uni-greifswald.de/webaugustus/examples/chr1to3.fa - This file may be used as a Genome file. It contains the first three chromosomes of Mus musculus from GenBank (modified headers).
http://bioinf.uni-greifswald.de/webaugustus/examples/traingenes.gb - This file contains 221 training genes for Mus musculus in GenBank format (including flanking regions).
1.6.4 - Automatic Data Insertion
You can insert some of these sample data sets by pressing the "Fill in Sample Data" button:
 |
After you click the "Start Training" button, the web server application first validates whether the combination of your input fields is generally correct. If you submitted unsupported input combinations, you will be redirected to the training submission form and an error message will be displayed at the top of the page.
If all fields were filled in correctly, the application is actually initiated. You will receive an e-mail that confirms your job submission and that contains a link to the job status page (if you supplied an e-mail address), and you will be redirected to the job status page.
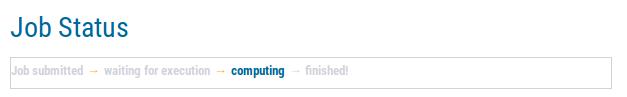 |
In the beginning, the status page will display that your job has been submitted. This means, the web server application is currently uploading your files and validating file formats. After a while, the status will change to waiting for execution. This means that all file formats have been confirmed and an actually AUGUSTUS training job has been submitted to our grid engine, but the job is still pending. Depending on waiting queue length, this status may persist for a while. Please contact us in case you job is pending for more than one month. Later, the job status will change to computing. This means the job is currently computing. When the page displays finished, all computations have been finished and a website with your job's results has been generated.
You will be notified by email when your job has finished - if you supplied an email address.
Since training AUGUSTUS is a very resource consuming process, we try to avoid data duplication. In case you or somebody else tries to submit exactly the same input file combination more than once, the duplicated job will be stopped and the submitter of the redundant job will receive information where to find the previous job status page.
You should automatically receive an e-mail in case an error occurs during the AUGUSTUS training process. The admin of this server is also notified by e-mail about errors. Please contact augustus-web@uni-greifswald.de if you want to find out what went wrong.
After job computations have finished, you will receive an e-mail (if you supplied an email address). The job status web page may at this point in time look similar to this:
 |
This page will should contain the files AutoAug.log and AutoAug.err. If no other files are displayed, you will find the reason for this behaviour in one of those files (e.g. if the creation of a sufficient number of training genes was not possible using the data that you supplied).
If training was successful, at least the files parameters.tar.gz and ab_initio.tar.gz should be displayed.
If no training gene structure file was supplied, training.gb.gz should be linked.
If a cDNA file was submitted, hints_pred.tar.gz should be shown.
To save files on your local system, click the file link with the right mouse button and select "Save Link As..." (or similar).
Unpack *.tar.gz archives locally on your system by typing tar -xzvf *.tar.gz into your shell. (You find more information about the software tar at the GNU tar website.)
Results of your job are deleted from our server after 180 days.
To view a real training output example, click here!
This file contains all logging messages of the AUGUSTUS training and prediction processed invoked by the AUGUSTUS training web server application. It is a plain text file, i.e. you should be able to open it with any text editor or even your web browser. In your own interest, you should check the AutoAug.log file for the number of training genes that were generated from your input files (except if you submitted a training gene structure file), for the number UTR training genes, and for gene prediction accuracy.
If your job ran successfully, the log file will contain a line that says 1 training set training.gb.train contains x sequences and y genes.. If the number of x is low, you should probably refrain from further using the produced parameter set. From our experience, you need at least a couple of hundreds of training genes in order to obtain good parameter sets. If UTR training was possible, the log file will contain similar information for the number of UTR training examples. Here, you also want to see a number that is at least a couple of hundreds - otherwise do not use the produced UTR parameters for gene prediction.
The log file also contains a line that says 1 The accuracy after optimizing without CRF-etraining is z. This accuracy value was obtained from a test gene set that was not used for training computed with the following formula:
3*nucleotide_sensitivity + 2*nucleotide_specificity + 4*exon_sensitivity + 3*exon_specificity + 2*gene_sensitivity + 1*gene_specificity)/15
Commonly observed values at this position range from 40 to 60 percent. If you obtain a very low value, this gives a strong indication that the obtained parameter set is not very useful for predicting genes accurately.
This file contains all error messages of the AUGUSTUS training and prediction processed invoked by the AUGUSTUS training web server application. It is a plain text file, i.e. you should be able to open it with any text editor or even your web browser. In your own interest, you should check the AutoAug.err file. If this file is not empty, something did go wrong and you probably shouldn't trust the results blindly - in case any were produced at all.
One frequently occuring error in the AutoAug.err file is the following:
The file with UTR parameters for train****** does not seem to exist. This likely means that the UTR model has not beeen trained yet for train******.
This error message tells you that no UTR parameters were trained for your species. If no other error messages are contained above the first UTR error message, the general results of your job are ok, you simply did not get UTR parameters and thus no predictions with UTR.
Further error messages (that e.g. lead to null results) are explained on our Help page.
This file contains the parameters that were trained for your species of interest based on the submitted input files. The AUGUSTUS parameter archive always contains the following files for your species (the term species will be replaced by the species name that you entered when submitting a training job):
- species_parameters.cfg
- species_metapars.cfg
- species_metapars.utr.cfg
- species_exon_probs.pbl.withoutCRF
- species_exon_probs.pbl
- species_weightmatrix.txt
- species_intron_probs.pbl
- species_intron_probs.pbl.withoutCRF
- species_igenic_probs.pbl
- species_igenic_probs.pbl.withoutCRF
If you want to use the parameters for predicting genes with a local AUGUSTUS installation, place the species parameter folder into your $AUGUSTUS_CONFIG_PATH species folder. Be aware that in general, our web server application does not optimize the CRF parameters. Therefore, do not run AUGUSTUS with CRF-flag with these parameters. UTR parameters have only been optimized for your species if this website contains an archive utr_pred.tar.gz. It does not make sense to locally predict genes with UTR if the UTR parameters were not optimized for your species.
If you want to run further predictions for your species of interest in other genomic sequences or with other hints on our web server application, you do not need to save the parameter archive locally for re-upload. Just remember the ID of your job and specify it on the prediction interface. You find your job's ID in the headline of this web site at "Training AUGUSTUS Job ..." or in the first message at "Training job ID: ... ".
This file contains generated training genes that were used to optimize AUGUSTUS parameters in genbank format. The file is compressed with gzip. To unpack it, type gunzip training.gb.gz in your shell. You find more information about gzip at gzip.org.
The files ab_inito.tar.gz and pred_hints.tar.gz are gene prediction archives. Their content depends on the input file combination.
Files that are always contained in gene prediction archives:
- *.gff - gene predictions in gff format
You find an example for AUGUSTUS prediction gff format at the AUGUSTUS prediction tutorial.
Files that may optionally be contained in gene prediction archives:
- *.gtf - gene predictions in gtf format
- *.aa - gene predictions as protein fasta sequences
- *.codingseq - gene predictions as CDS DNA fasta sequences
- *.cdsexons - predicted exons in DNA fasta sequences
- *.mrna - predicted mRNA sequences (with UTRs) in DNA fasta sequences
- *.gbrowse - gene prediction track for the GBrowse genome browser
[1] Haas, B.J., Delcher, A.L., Mount, S.M., Wortman, J.R., Smith Jr, R.K., Jr., Hannick, L.I., Maiti, R., Ronning, C.M., Rusch, D.B., Town, C.D. et al. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res, 31, 5654-5666.
[2] Kent, W.J. (2002) BLAT—The BLAST-Like Alignment Tool. Genome Res, 12, 656-664.
[3] Keller, O., Odronitz, F., Stanke, M., Kollmar, M., Waack, S. (2008) Scipio: Using protein sequences to determine the precise exon/intron structures of genes and their orthologs in closely related species. BMC Bioinformatics 9, 278.
[4] Stanke, M., Diekhans, M., Baertsch, R., Haussler, D. (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, doi: 10.1093/bioinformatics/btn013